執筆:スクマラン サティシュ・滝本淳一(山形大学)
きめ細かい記述は、系の微視的な詳細を用いた記述を示します。 粗視化記述は、その微視的な詳細の一部を「平均化」されたことを表します。
コンピュータ シミュレーションの目標の一つは、数学的モデルを用いて現実系の静的・動的物性を研究することです。 コンピュータプロセッサは継続的に高速化され、またその計算力は徐々に利用しやすくなっています。 しかし、巨視的系は通常、アボガドロ数程度の副単位(原子、分子など)で構成されています。 多くの巨視的系では、対象となる現象が複数の空間スケールおよび時間スケールで発生し、非常に遅い運動と長い緩和時間を伴います。 巨視的系のこれらの特性により、高分子系の動力学・レオロジー・成形加工、半結晶性高分子の階層構造及び結晶化過程 (Hierarchical structure of and the crystallization process in semi-crystalline polymers)、ミクロおよびマクロ相分離 (Micro and Macro phase separation)、界面活性剤の自己集合(Surfactant Self-Assembly)、生体膜 (biomembranes) およびタンパク質のフォールディング (Protein Folding)、などの多くの現象を全原子論的アプローチで解明することは、ほとんど不可能になります。 粗視化を使用してさまざまな空間および時間スケールで発生する現象を適切に記述できる階層的モデルを開発するのは、複数スケールで発生する複雑な現象を理解するための 1 つの経路になります。さらに、粗視化モデルを使用したシミュレーションは、連続体スケールでの構成方程式に必要な入力パラメータを直接提供し、原子スケールと連続体スケールを結びつけます。
以下では、まず粗視化の背後にある基礎考え方を簡単に紹介します。 続いて、高分子系に現在使用されている粗視化アプローチについて具体的な例に基づいて概説します。 言うまでもなく、粗視化アプローチと応用例の広範な説明は、この記事の範囲を超えています。興味のある読者は種々の合成高分子、生体分子、およびその他の種々のソフトマターに適用される粗視化アプローチの詳細について文献
1)-7) で参照することをお勧めします。
測定できること
絡み合い高分子の動力学・レオロジー / 半結晶性高分子の階層構造及び結晶化過程 / ミクロ・マクロ相分離構造・動力学 / 界面活性剤の自己集合での構造形成及びその動力学
一般的なキーワード
粗視化 (Coarse-graining) / 解像度の粗さ / 記述レベル / 粗視化ポテンシャル / iterated Boltzmann inversion / 平均力ポテンシャル
原理
1.粗視化とは
粗視化の基礎的な考え方は、系の全自由度のうち、究明の対象の現象を適切に記述するために必要な自由度のみを保つことです。 その結果、モデルの自由度数が減少し、多くの場合、大幅かつ自然にモデルが単純化されます。 ただし、単純化すると、元のモデルに存在する化学的詳細が徐々に失われます。 したがって、原子スケールでの現象を記述することはもはや不可能ですが、粗視化モデルを使用することで、より長い空間および時間スケールの現象の究明が可能になります。基礎となる化学構造の影響も平均的には考慮できます。一般に粗視化モデルでは、どの自由度を含めるかの選択が重要になります。 ただし、これは必ずしも簡単ではなく、多くの場合、物理的な洞察が必要です。系の粗視化記述の重要な特徴は、対象の系の微視的情報だけに基づいて粗視化モデルを構築しており、それ以外の恣意的な操作を行っていないことです。 これが、粗視化記述が究明の対象系の有効の記述であると考えられる基本的な理由です。記述のきめ細かさはもちろん低下しますが、長距離・長時間では系の忠実な記述が可能になることは、他の抽象化方法に比べて粗視化を区別する 1 つの重要な点です。
具体的な例を考えてみましょう。 ほとんどの読者は平衡熱力学はご存知と思います。 しかし、平衡熱力学は多粒子系の粗視化記述の優れた例であることは、あまり知られていないかもしれません。 多粒子系の挙動は、統計力学を使用して研究できます。 統計力学では、系の取り得る個々の状態の完全な記述は、その微視的状態 (microstate) で与えられます。 たとえば、古典的統計力学では、多粒子系の各粒子は 6 つの数値(3 つの空間位置と 3 つの運動量)で記述されます。 したがって、N 粒子で構成された系の微視的状態を記述するには、6N 個の数値が必要になります。
これとは対照的に、平衡熱力学における単一成分系の巨視的状態 (macrostate) を定義するには、体積、圧力、化学ポテンシャル、そしておそらく最も重要な温度などのいくつかの巨視的物理量で十分です。ここで重要な点は、各巨視的状態には数多くの微視的状態が対応するということです。 明らかに、これは、多粒子系を記述するために使用される変数の数が大幅に減少することを意味します。 これをよりよく理解するために、温度の概念に焦点を当ててみましょう。
統計力学では、温度は系内粒子の平均運動エネルギーを使用して定義されています。 言い換えれば、温度は系内の全粒子の運動を集約した表現になります。 信じられないことに、系の温度を上記の他のいくつかの巨視的変数とともに知ることで、将来の熱力学的挙動を予測することが可能になります。 個々の粒子の速度を知る必要はありません。 これは、粗視化の驚くべき有用性を示しており、その重要性を表しています。粗視化には通常、複数の微視的状態にわたる積分が必要になります。 前述したように、温度は系内の粒子ごとの平均運動エネルギーを使用して定義できます。 このような平均が最も単純な例ですが、より複雑な巨視的物理量も、究明の対象の現象の記述・理解に役立つことが証明されています。
2.粗視化戦略
原子スケールでは、基礎原理にしっかりと基づいた戦略を使用して、量子力学モデルから古典力学モデルまでの粗視化を実現できます。 これらの戦略は、必要に応じて一般化できます。しかし、より大きな空間および時間スケールでは、基礎原理に基づいた一般的な戦略での粗視化は不可能になり、適切な粗視化戦略は究明の対象の特定系に応じて異なります。 さらに、粗視化前の微視的モデル(たとえば全原子モデル)と粗視化モデルの対応を定義する関数(mapping)も、究明したい現象に依存し、一意ではありません。 現在、高分子系には、きめの細かいモデルのシミュレーションを利用して得られるシステムの構造相関と熱力学的な物性を適切に再現できる粗視化戦略がいくつか存在します。 開発した粗視化ポテンシャルを別の系の研究に再利用するのは依然として問題ですが、特定物性の一致を体系的に改善するための道筋は知られています。 ただし、これは粗視化モデルの動力学には当てはまらず、かなり異なる課題が生じます。 通常、粗視化モデルは、基となる微視的モデルと比べて、大幅に加速された運動を示します。 このような加速された運動は、粗視化過程における人工的な効果 (artifact) であると考えられています。 高分子系の構造・熱力学物性だけでなく、その階層的動力学も適切に再現できる粗視化戦略の開発は、現在活発な研究分野です
7)。 粗視化モデルの動力学は微視的モデルの動力学にどのように関連するか、多くの不明点がまだ残っています。
3.体系的粗視化
3-1 粗視化ポテンシャル
おそらく、高分子系の粗視化に最も広く使用されている戦略は、全原子モデルでのいくつかの原子を一つの単位(粗視化粒子)にグループ化することです。高分子鎖固有の長さおよび時間スケールの階層記述能力を維持しながら、計算コストを大幅に削減することができます(図 1 を参照)。 多くの場合、前述の戦略の最初のステップは、ユナイテッドアトム(united-atom)モデルの開発です。 「ユナイテッドアトム」とは、炭素原子とそれに共有結合しているすべての水素原子が単一の擬似原子にグループ化されていることを指します。 重要なのは、擬似原子間のポテンシャルを導出しパラメータ化することです。これは、全原子シミュレーションを使用するか、実験結果を再現するためのパラメータマッチングによって達成されます。 脂肪族炭化水素のユナイテッドアトムモデルの例は、GROMOS96 力場
8)です。 ユナイテッドアトムのみでなく、いくつかの原子または分子を単一の粗視化単位にグループ化することができます。 このようなアプローチの一例は、マルティーニ力場
9) です。 マティーニ力場のアプローチは元々は生体分系用に開発されましたが、高分子系などのソフトマターにも応用できるようになりました
9)。
.jpg) 図1 ポリエチレンの段階的粗視化
図1 ポリエチレンの段階的粗視化
前述の粗視化戦略は、粗視化単位間のポテンシャルの導出に大きく依存しています。 2 体の局所構造の一致を確保することに限定すると、粗視化単位間の2 体間非結合ポテンシャルを計算するアプローチが存在します。 この目的で最も広く使用されているアプローチは、iterative Boltzmann inversion
10)です。 このアプローチでは、動径分布関数 (radial distribution function, RDF) のボルツマン逆変換によって得られる距離
r だけ離れた 2 つの種
i と
j の分子間の平均力ポテンシャル (Potential of Mean Force) から開始します。
ここで、
gij(
r) は粗視化単位の 2 つの種
i と
j の間の目標RDF、
kB はボルツマン定数、
T は系の温度です。式(1)を用いて計算したポテンシャル
.jpg)
は、無限希釈領域では正確ですが、高密度系の場合はは不適切であることが知られています。 言い換えれば、
を使用したシミュレーションでは目標RDF を再現することはできません。 ただし、初期推定として
を使用し、式(2)を用いることでポテンシャルの精度を向上させることができます。
ここで、
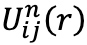
は反復
n後の種
i と
j 間の有効的2体ポテンシャルです。 式(2)を使用した反復手順は、有効的2体ポテンシャルから生成されたRDFが目標RDFに所望の数値精度に収束するまで繰り返されます。
3-2 粗視化動力学
上記の粗視化ポテンシャルにより粗視化前後での系の構造は統計的に同様となります。しかし、粗視化粒子の運動の記述は別途考えなければなりません。
Mori-Zwanzig理論
11),12) に基づいた射影演算子 (Projection Operator) 法は、粗視化表現に適した動的変数を抽出するための一般戦略です。 この戦略では、粗視化変数以外の自由度を排除することで、粗視化変数の運動方程式を導出できます。 結果として得られる運動方程式は一般化されたランジュバン型であり、散逸力に対応する決定論的な記憶項と変動 (ランダム) 力に対応するノイズ項を備えています。 射影演算子方法については
13)を参照してください。
自由度を削減するための射影演算子法の背後にある主な考え方は非常に一般的であり、原理的にはさまざまな系に適用できます。 ただし、結果として得られる一般化ランジュバン方程式は、おそらく比較的に単純な系以外は、かなり複雑になります。 したがって、得られた方程式を実際に使用する前に、追加の近似が必要になります。 粗視化に射影演算子アプローチを利用した最近の取り組みの概要は、
7)に見つかります。
4.経験的粗視化
4.1 実験的に確立された普遍性に基づく粗視化モデリング
長い空間および時間スケールでの高分子系のいくつかの特徴的な物性は、高分子が長い鎖状であることの直接的な結果です。 このような物性は、ポリマーの特定の化学構造に特有のものではく、様々な高分子種類で観察できます。 したがって、特定の化学構造の全原子モデルから始めて粗視化モデルを開発する必要はありません。 その代わり、対象現象を支配すると考えられる特徴だけを残しながら、高い計算効率持つモデルを構築できます。 具体的な例を考えてみましょう。
絡み合い高分子液体では、高分子鎖の緩和時間は原子時間スケールよりも数桁ほど長いです。 コンピューター シミュレーションを使用してこのような液体の長時間の動力学を研究するため、次の特徴を組み合わせたモデルを開発できます。
- 液体のような局所構造: 液体の局所構造は、主に、近距離で作用する粒子間の強い反発力によります。 したがって、レナード-ジョーンズポテンシャルの反発部分のみが働く粒子の液体が考えられます。
- 鎖の接続性と柔軟性: 高分子鎖をシミュレーションするには、液体の粒子をバネで繋ぐ必要があります。 このような鎖は本来柔軟性があり、温度に応じてブラウン運動によって形状を変化させることができます。 高分子鎖の柔軟性は、曲げポテンシャルを使用することでより正確に調製できます。
- 高分子鎖の相互非すり抜け性: 排除体積効果により、2つの高分子鎖は相互にすり抜けることはできません。この特徴は、絡み合いによる現象・物性を再現するために必要です。非すり抜け性は、2 つの鎖のすり抜けに必要なエネルギーを法外に高くすることで実現できます。 ポリマー モデルで実装する 1 つの方法は、結合長がすり抜けが可能な長さまで決して伸びない結合ポテンシャルを使用することです。
上記の特徴を組み込んだモデルが、Kremer と Grest によって開発されました
14)。 このモデルは、絡み合い高分子の動力学の研究に広く使用されており、理解に大きく貢献しています。 詳細と参考文献は、
1),4),15)に見つかります。
4.2 具体例: 絡み合い高分子の動力学検討に使用する階層的モデル
ここで、絡み合い高分子の動力学とレオロジーの記述に広く使用されている階層モデルの概要を説明します (図 2 を参照)。より詳しい説明は
15)を参考してください。
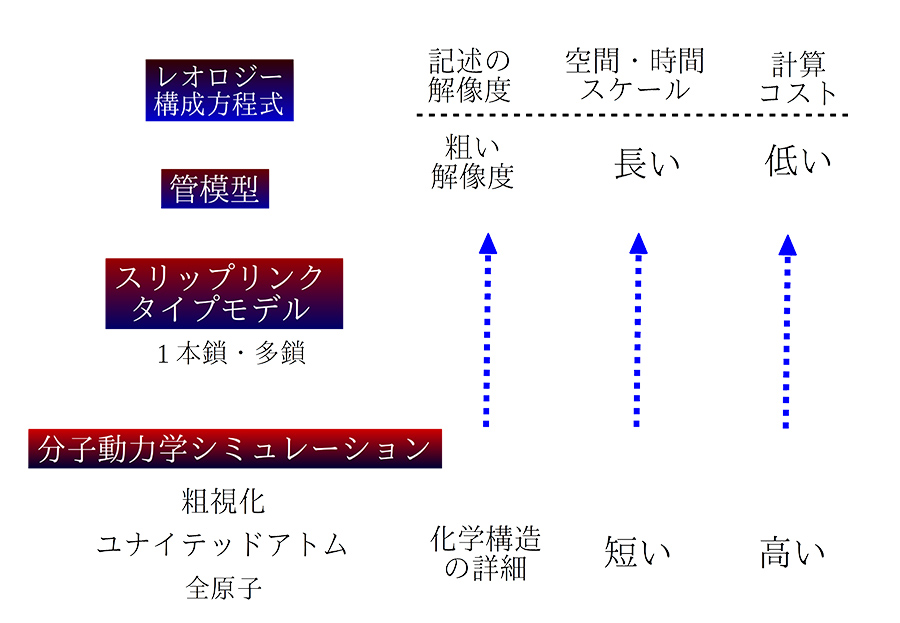 図2 絡み合い高分子の動力学に使用する階層モデル
図2 絡み合い高分子の動力学に使用する階層モデル
全原子モデルには、特定の高分子の化学構造の詳細がすべて含まれています。 このようなモデルは、結合回転や局所的な原子運動などの現象を観察および理解したい場合に必要です。 より長い空間と時間スケールの場合、粗視化分子動力学シミュレーションが採用され、重要な洞察が得られました
14),1),4)。 より長いスケールでは、管模型や、スリップ スプリングおよびスリップ リンク モデルが効果的であることが証明されています
15)。 これらのモデルにはシングルチェーン バージョンとマルチチェーン バージョンがあります。 さらに、モデルによっては、高分子鎖の粗視化のレベルがかなり異なる場合があります。 巨視的なレオロジー現象の記述には、レオロジー構成方程式の使用が適切です。 このような構成方程式は、高分子の成形加工に該当する流動計算に利用できます。
5.ソフトウェアパッケージ
上記で説明したように、粗視化はシミュレーションは単なる手法でなく、さまざまな長さと時間スケールに適したモデルを開発する際に役立つ思考法を指します。 粗視化モデルは多くの場合、検討対象の特定現象に非常に特化しているため、汎用的に使用できるソフトウェアの推奨は困難ですが、表1に多く使われている代表的なソフトを示します。
表 1: ソフトウェアパッケージ
参考文献
1) Baschnagel, J. Binder, K. Doruker, P. Gusev, A. A. Hahn, O. Kremer, K. Mattice, W. L. Müller-Plathe, F. Murat, M. Paul, W. Santos, S. Suter, U. W., Tries, V. "Bridging the Gap Between Atomistic and Coarse-Grained Models of Polymers: Status and Perspectives" in Viscoelasticity, Atomistic Models, Statistical Chemistry, Advances in Polymer Science 152, 41–156 (2000).
2) Peter, C. Kremer, K. "Multiscale simulation of soft matter systems – from the atomistic to the coarse-grained level and back", Soft Matter 5, 4357-4366 (2009).
3) Saunders, M. G. Voth, G. A. Annual Reviews of Biophysics 42, 73-93 (2013).
4) Dhamankar, S. Webb, M. A "Chemically specific coarse-graining of polymers: Methods and prospects", Journal of Polymer Science, 59, 2613-2643 (2021).
5) Jin, J. Pak, A. J. Durumeric, A. E. P. Loose, T. D. Voth, G. A. "Bottom-up Coarse-Graining: Principles and Perspectives", Journal of Chemical Theory and Computation, 18, 5759–5791 (2022).
6) 基礎高分子科学第2版 第10章 高分子学会編 東京化学同人(2020).
7) 基礎高分子科学演習編第2版 第10章 高分子学会編 東京化学同人(2023).
8) Schuler, L.D. Daura, X. van Gunsteren, W.F. "An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase", Journal of Computational Chemistry 22, 1205-1218 (2001).
9) Alessandri, R. Grünewald, F. Marrink, S.J. "The Martini Model in Materials Science", Advanced Materials 33, 2008635-1-2008635-20, (2021).
10) Reith, D. Pütz, M. Müller-Plathe, F. Journal of Computational Chemistry, 24, 1624-1636 (2003).
11) Zwanzig, R. "Ensemble Method in the Theory of Irreversibility", Journal of Chemical Physics, 33, 1338–1341 (1960).
12) Mori, H. "Transport, collective motion, and Brownian motion", Progress of Theoretical Physics, 33, 423-455 (1965).
13) Zwanzig, R. "Nonequilibrium Statistical Mechanics", 1st edition, Oxford University Press (2001), 143-168.
14) Kremer, K. Grest, G. S. "Dynamics of entangled linear polymer melts: a molecular-dynamics simulation", Journal of Chemical Physics 92, 5057–86 (1990).
15) Masubuchi, Y. "Simulating the Flow of Entangled Polymers", Annual Review of Chemical and Biomolecular Engineering, 5, 11-33 (2014).
16) 増補版 高分子材料シミュレーション: OCTA活用事例集 新化学技術推進協会編 化学工業日報社(2017).
17) Thompson, A. P Aktulga, H. M. Berger, R. Bolintineanu, D. S. Brown, W. M. Crozier, P. S. in 't Veld, P. J. Kohlmeyer, A. Moore, S. G. Nguyen, T. D. Shan, R. Stevens, M. J. Tranchida, J. Trott, C. Plimpton, S. J. "LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales", Comp Phys Comm, 271, 108171-1-108171-34 (2022).
18) Weik, F. Weeber, R. Szuttor, K. Breitsprecher, K. de Graaf, J. Kuron, M. Landsgesell, J. Menke, H.Sean, D. and Holm C. “ESPResSo 4.0 – an extensible software package for simulating soft matter systems”, The European Physical Journal Special Topics 227, 1789–1816 (2019).
19) Brooks, B. R. Brooks III, C. L. Mackerell, A. D. Nilsson, L. Petrella, R. J. Roux, B. B. Won, B. Archontis, G. Bartels, C. Boresch S. Caflisch, A. Caves, L. Cui, Q. Dinner, A. R. Feig, M. Fischer, S. Gao, J. Hodoscek, M. Im, W. Kuczera, K. Lazaridis, T. Ma, J. Ovchinnikov, V. Paci, E. Pastor, R. W. Post, C. B. Pu, J. Z. Schaefer, M. Tidor, B. Venable, R. M. Woodcock, H. L. Wu, X. Yang, W. York, D. M. , Karplus, M. "CHARMM: The Biomolecular simulation Program", Journal of Computational Chemistry 30, 1545-1615 (2009).
20) Lu, L. Izvekov, S. Das, A. Andersen, H. C. Voth, G. A. "Efficient, Regularized, and Scalable Algorithms for Multiscale Coarse-Graining", Journal of Chemical Theory and Computation, 6, 954–965 (2010).
.jpg)
.jpg)