執筆:折本 裕一(九州大学)
量子化学計算は、量子力学に基づいて対象系の電子の挙動とその関連特性を調べる手法
量子化学計算は、量子力学のシュレーディンガー方程式に基づいて対象系の電子の挙動を調べる理論手法です。電子に関連した様々な物性(導電性、磁性、光学特性等)や現象(化学反応等)を調べることができます。その中心的理論である分子軌道法において、結果として得られる分子軌道や軌道エネルギーなどを解析することで物性・現象の原因を電子レベルで探究することができます。計算コストが大きいため、通常は比較的小さな分子系について用いられる量子化学計算ですが、高分子への適用においても様々なアプローチが存在します。
測定できること
電子状態 / 安定構造 / 結合エネルギー / 導電性 / 磁性 / (非線形)光学特性 / 光・化学反応 / 遷移状態 / 励起状態 / 分子振動
原理
はじめに
量子化学計算は、今やソフトウェアも充実し、研究者にとって身近なものとなりました。計算機の進歩も目覚ましく、デスクサイドのコンピュータ上でも、ある程度の計算は実行できます。ただし、相手が高分子だと、状況は一変します(
図1)。計算コストが大きい量子化学計算にとって、系に含まれる原子数が多すぎるのです。高分子が完全な周期性をもっていれば周期境界条件を用いた確立された計算方法がありますが、現実には多くのタンパク質が複雑なアミノ酸配列を持ち、また、精密合成した材料系ポリマーも多様なコンフォメーションや末端構造のために、結局は非周期系です。系を丸ごと計算できれば良いのですが、量子化学計算の適用は、高性能サーバや最先端のスパコンを用いても容易ではありません。本解説では、量子化学計算の特徴とともに、高分子研究に利用する際の指針と注意点について紹介します。
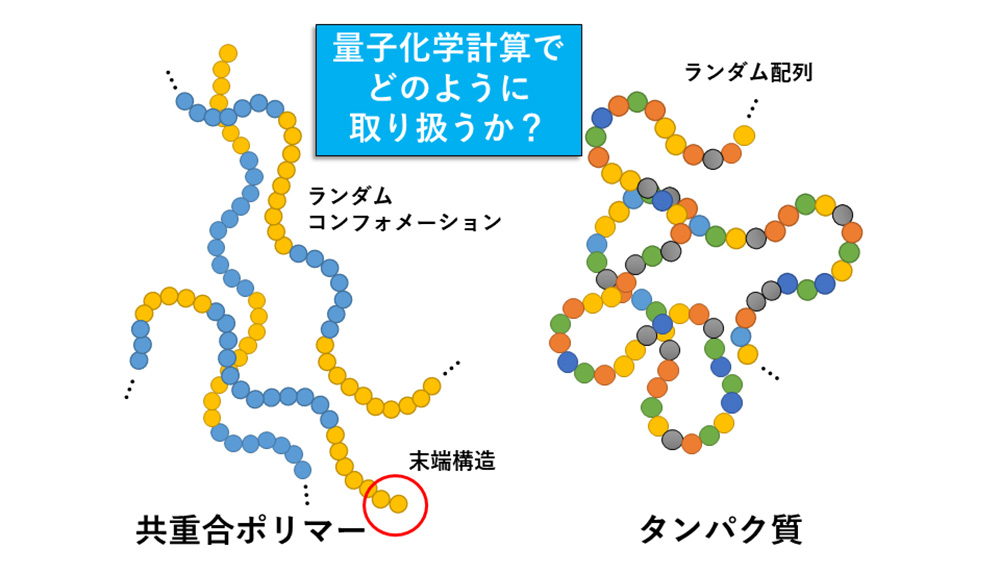 図1 高分子の例
図1 高分子の例量子化学計算から分かること
量子化学は「電子」の挙動を調べる理論です。シュレーディンガー方程式ĤΨ=EΨ(
式1)に基づいて対象系の電子状態やエネルギーが得られます(分子の場合、Ĥ、Ψ、Eはそれぞれ多電子系のハミルトニアン、波動関数、エネルギー)。電荷分布や双極子モーメントといった基礎情報をはじめ、導電性・磁性・(非線形)光学特性などの電子に関連した特性を調べることができます。安定構造、結合エネルギー、分子振動のほか、実験では短寿命のため調べることの難しい遷移状態、励起状態、結合生成・解離を伴う化学反応なども計算から分かります。実験値との対応では、UV-Vis吸収スペクトルやNMR化学シフト、赤外・ラマン振動スペクトルなどを予測できます。あらゆる物性・現象が電子に関係するという意味では、その守備範囲は広いと言えます。
量子化学計算の注意点
一方で、量子化学計算は絶対零度下の理論であることに注意が必要です。近似的に温度効果を考慮し自由エネルギーを見積もることも出来ますが、多くの場合、温度を含まない「全エネルギー」によって系の安定性を議論します。量子化学計算の構造最適化の手続きでは、初期構造からポテンシャルエネルギーを下って局所的な安定構造を見出します。広範囲により安定な構造を探索するには、通常は複数の初期構造から最適化を行ない、エネルギーの最も低い安定構造を決定します。これが、検討した中での最安定構造という位置づけになります。無数のコンフォメーションを取りうる高分子ではこのような安定構造を決めること自体が課題です。また、計算対象分子の周囲環境については、何も設定しなければ真空中の孤立分子の計算になります。環境を考慮したいときは、実際に周りに分子を配置するか、あるいは近似的に溶媒効果を考慮する方法もあります。
量子化学計算の種類
一般に量子化学計算と言えば、化学分野で発展してきた分子軌道法(molecular orbital(MO)法)を指します。
式1は、多電子系の場合は多体問題のため厳密には解けません。そこで、注目する1つの電子が他の電子が作る平均場中を運動する「軌道」(1電子波動関数)という近似概念が導入されました。分子中の電子運動の振舞いを表す関数は特に分子軌道(MO、
φi)と呼び、
φiによって多電子波動関数Ψを構成します。MO法の中心的手法はハートリー・フォック(HF)法と呼ばれます(
図2)。1電子軌道近似によって取り込めていない電子どうしが避け合う効果(電子相関効果)を考慮するため、HF法を超えたMP摂動法、配置間相互作用(CI)法、クラスター展開法などのポストHF(post-HF)法が提案されています。電子相関効果は、例えば、ファンデアワールス力などの弱い相互作用の評価に欠かせません。経験的パラメータを用いていないHF法やpost-HF法は、非経験的(
ab initio)MO法と呼ばれます。
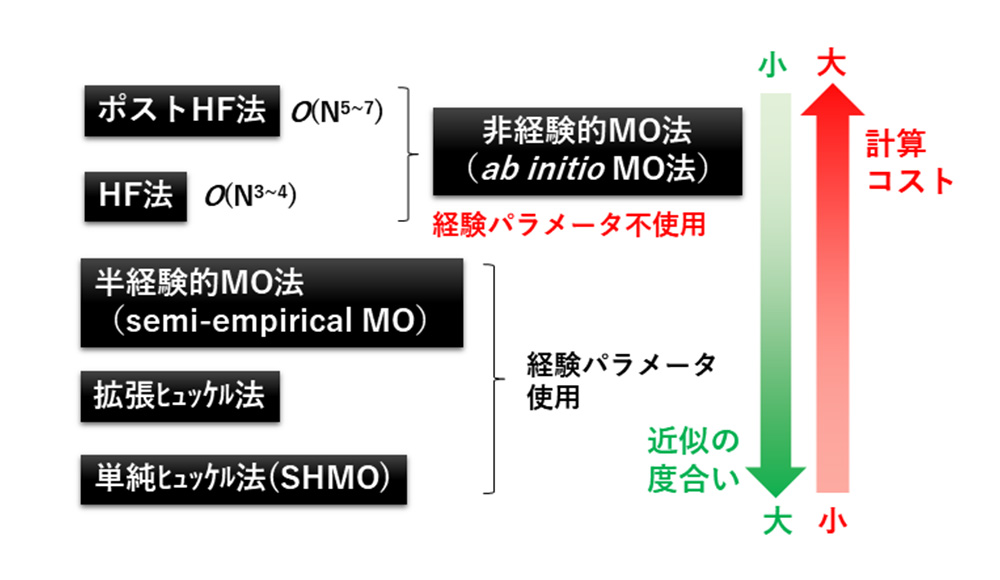 図2 量子化学計算の種類
図2 量子化学計算の種類
量子化学計算は、膨大な積分計算と行列固有値計算から成り立っていて、計算コストは系のサイズNに対して指数関数的に増加します(厳密には後述の基底関数の数に依存)(
図3)。計算コストとは、演算時間だけでなく、計算中の情報を保存するためのメモリやHDD容量をも含みます。HF法ではNの3~4乗オーダーで計算コストがかかり、
O(N
3~4)と表現します。post-HF法ではさらにコストが膨大で
O(N
5~7)にもなります。この指数関数的なコスト増大が、量子化学計算の高分子への適用を困難にしています。
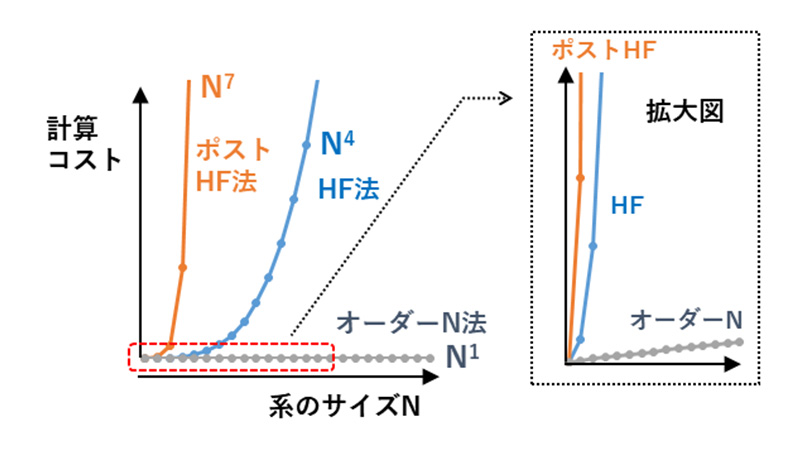 図3 分子サイズと計算コスト
図3 分子サイズと計算コスト
限られた計算資源の中で量子化学計算を実行するため、多くの近似手法が提案されてきました。積分計算に経験的パラメータを導入する半経験的MO法や、π電子のみ考慮する単純ヒュッケルMO法(simple Hückel MO(SHMO)法)などがあります(
図2)。パラメータ依存性に注意する必要がありますが、近似が粗いほど大きな系が計算できるため、高精度を求めない場合は利用しやすい方法です。
電子状態を得るための別の手段として、固体・表面の計算に利用される物理分野の密度汎関数法(density functional theory (DFT)法)があります。第一原理計算とも呼ばれ、原子に局在した基底ではなく、平面波を基底としたアプローチです。その後、DFT法に後述の原子局在基底関数が導入され、化学分野における分子系の計算にも広く使われています。電子密度の汎関数で与えられるコーンシャム方程式を解きますが、電子相関効果は汎関数内に記述され、HF法と同程度のコストで電子相関を考慮できます。汎関数(B3LYP、B3PW91など)の選択には経験的側面があり、半経験的手法の一面も持っています。
分子軌道と基底関数
分子中の電子の運動を記述するMOについて、各原子付近ではその原子軌道(atomic orbital (AO))に似ているという発想に基づき、MOを既に形の分かっているAOの線形結合
φi = Σ
rC
irχ
r(
式2)で表します(
φi はi番目のMO、χ
rはr番目のAO)。これをLCAO(linear combination of AOs)近似と呼び、全エネルギーが最小になるように変分的にMO係数C
irを求め
φiを決定します。同時に、各MOに対応した軌道エネルギーε
iが得られます。
式2の既知関数群{χ
r}は基底関数系(basis set)と呼ばれ、計算時に指定します(3-21G、6-31+G(d,p)、aug-cc-pVTZなど)。大雑把に言えば、basis setに含まれる関数が多いほど
式2の表現力が上がり高精度と言えますが、同時に
図3のNが増えるため計算コストが急激に増大します。また、目的別に様々な基底関数が存在するため、基底関数系の増強の仕方は一通りではありません。基底関数系を適切に選択しないと、関数の数が増えても精度向上につながらないことがあります。また、HF法の枠内で基底関数をいくら増やしても限界となるHF極限の問題もあります。
MOの電子占有
線形代数により、使用した基底関数の数だけMOが得られます。ここで、計算対象分子が N
e個の電子を持っているとします。基底状態のとき、パウリの排他原理に従って軌道エネルギーε
iの低いMOから順に、上向き・下向きスピンをもった電子が2個ずつ、N
e/2番目のMOまで占有します(
図4)。電子占有されたMOのうち最もエネルギーの高い軌道がHOMO(highest occupied MO)、電子が入っていない空軌道のうち最低エネルギーのものがLUMO(lowest unoccupied MO)です。HOMOの軌道エネルギーは高いため、そこにいる電子は外部の求電子試薬分子のLUMOに移動しやすく、一方、エネルギーが低いLUMOは求核試薬分子のHOMOから電子を受け取りやすい軌道です。化学反応に関与するHOMO, LUMO付近の軌道をフロンティア軌道と呼びます。
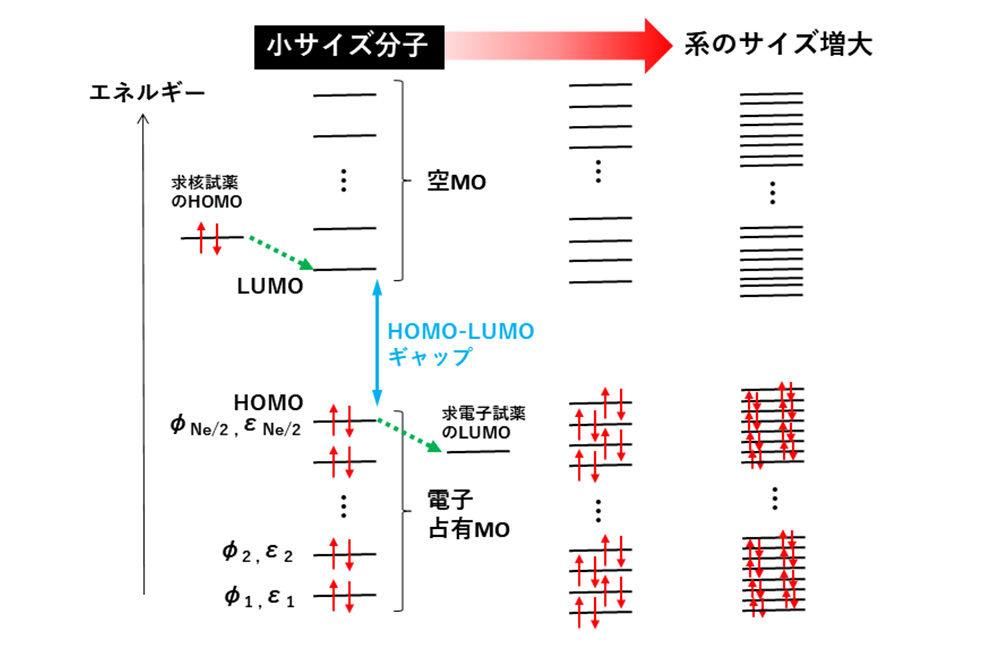 図4 MOの電子占有と系サイズ依存性
図4 MOの電子占有と系サイズ依存性はSHMO法から得たナフタレンのπ性を持ったHOMO及びLUMOです。SHMO法では炭素原子のπ電子のみ考慮するため、式2は分子面に垂直な2p軌道のみで線形展開します。MO係数の符号と大きさは、丸の色とサイズで表現しています。隣接したp軌道間の符号関係について(
図5(b))、同じ符号のとき同位相と言ってその部分は安定化に寄与し、異符号のとき逆位相と呼び、その部分は不安定化に寄与します。
図5(a)において、隣接p軌道間の逆位相(図中の破線位置)を数えると、HOMOよりもLUMOの方が多く、結果的に不安定な軌道として現れることに対応しています。MO係数の大きさは、HOMO、LUMOともにα位(2, 5, 7, 10の炭素位置)が他の部位よりも大きく、HOMOに対する求電子反応、LUMOに対する求核反応の両方についてα位が試薬分子に攻撃されやすいことを示しています。
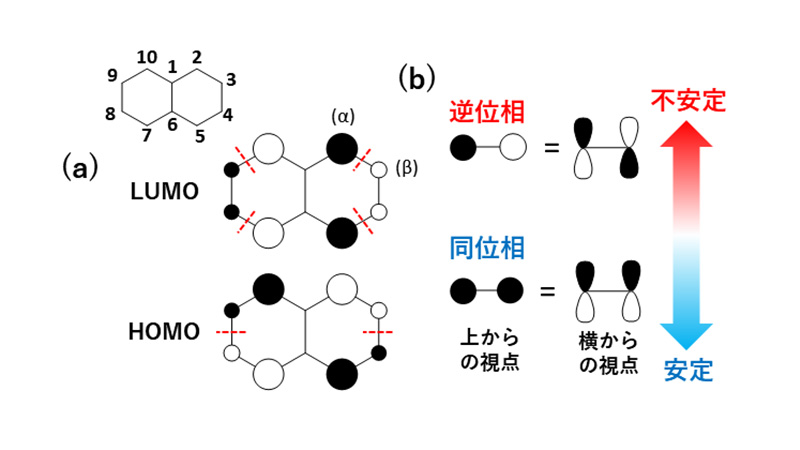 図5 (a)単純ヒュッケル法によるナフタレンのMO(破線は逆位相位置), (b) 隣接p軌道間の位相関係
図5 (a)単純ヒュッケル法によるナフタレンのMO(破線は逆位相位置), (b) 隣接p軌道間の位相関係高分子のMO
小分子の軌道エネルギーε
iはとびとびですが、高分子を想定して分子サイズを大きくしていくとMOのエネルギー間隔が徐々に狭まり、ほぼ連続したバンドに見えます(
図4)。
図6はSHMO法によるパラフェニレンオリゴマー(重合度n=5)のHOMOとLUMOです。ここではオリゴマーを用いますが、高分子を想定しても同様の説明が可能です。図下に、エネルギーが2つずつ縮重したベンゼンのHOMO, LUMOのうちの一方を示します。HOMOでは、5つのベンゼンHOMOが互いに逆位相の関係でつながっています。逆位相のため不安定化した結果、n=5のHOMOを形成しています。同様に、LUMOは5つのベンゼンLUMOがすべて同位相でつながり安定化した結果であることが分かります。このように高分子のMOは構成モノマーのMOに基づいて説明可能であり、分子特性・現象の原因解明に有用な情報を与えてくれます。
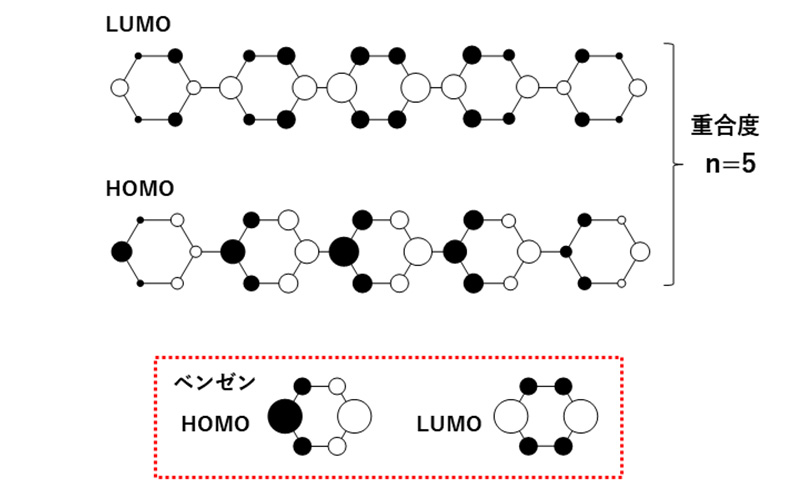 図6 パラフェニレンオリゴマーのMO
図6 パラフェニレンオリゴマーのMO
具体例として、ポリエンの結合交替(
図7下の長/短結合長の繰り返し)についてMOから説明します。
図7上はSHMO法から得たポリエン(重合度n=8)のHOMOとLUMOです。
図6と同様に考えると、ポリエンのHOMOはモノマーであるエチレンHOMOが逆位相で結合し、LUMOはエチレンLUMOが同位相で結合しています。さて、ここでポリエンのHOMOを見ると、a, b, c位置ではC-C長を短くして同位相関係を強めて安定化しようとし、d, e, f位置ではC-C長を長くすることで逆位相関係を弱めてこちらも安定化しようとします。これはパイエルス不安定性の一種と解釈でき、図下の結合交替を引き起こします。この構造変化はLUMOにとっては逆位相が近づき、同位相が離れるため、逆に不安定化につながります。結合交替を起こすことで系がより安定になることを説明するには、本来、全ての電子占有MOを考える必要がありますが、HOMO/LUMOの寄与が最大であることが摂動論で証明されているため、簡単のためこれらの軌道だけで説明を行いました。結合交替はどれだけ分子鎖を伸ばしても同様に起こります。これにより、両端を持った有限系のままいくら鎖長を伸ばしてもHOMOとLUMOは等価にならずHOMO-LUMOギャップが残ります。一方、後述のように無限周期系に拡張すると、C-C間が等間隔の場合はHOMO/LUMOの区別が完全になくなり縮重しますが、占有MOレベルを下げて安定化するよう結合交替を起こし、そのためポリアセチレンは導電性を持たないとされています。
本解説では、量子化学計算を紹介する入り口としてSHMO法を多用していますが、
Ab initio MO法などではより複雑になるものの、基本は同じです。SHMO法はコンピュータが無くても紙と鉛筆で計算できます。量子化学計算を学ぶ第一歩としておすすめします。
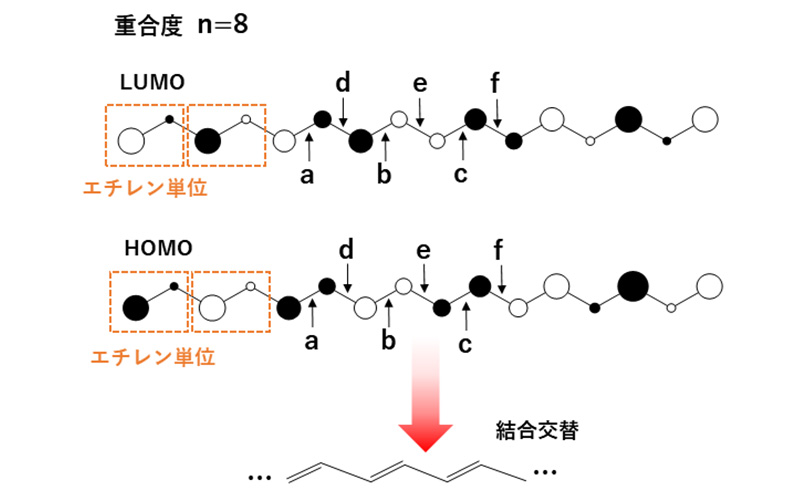 図7 ポリエンのMOと結合交替
図7 ポリエンのMOと結合交替量子化学計算の高分子への適用
量子化学計算をどのように高分子研究に活かすか、いくつか指針を示します。
1)計算モデルの切り出し
「はじめに」で述べた高分子を丸ごと…と矛盾しますが、計算できるサイズまで系を小さくするアプローチは、常套手段としてよく使われます(
図8)。高分子の調べたい部分を中心にモデルを切り出し、水素原子等で結合切断部をキャップします。新たに生じた末端が注目部位の電子状態になるべく影響しないよう、可能な限り大きく切り出したいところですが、計算コストも気になります。モデルサイズ(末端の影響)、計算レベル、計算コストの適切なバランスを、時に試行錯誤を行いながら適用可否を判断することになります。
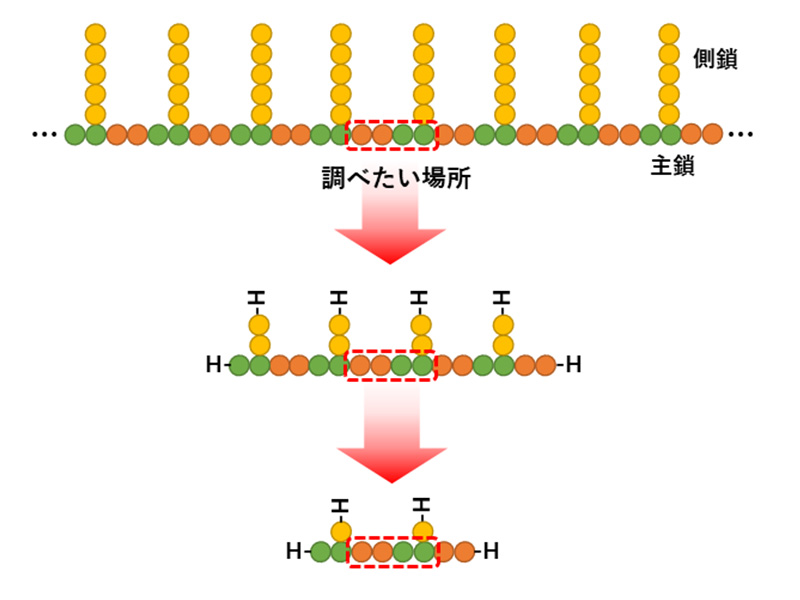 図8 計算モデルの切り出し
図8 計算モデルの切り出し
高分子を末端のない完全周期系として扱うアプローチです。例えばSHMO法において
図9(a)のようにN番目のサイトを1番目に接続した上でN→∞とすることで、無限周期系とみなせます。
簡単な例でMOに与える末端の影響を見てみます。
図9(b)はSHMO法による直鎖状と環状ポリエン(重合度n=5)の最低エネルギーのMO
φ1です。どちらのMOもエチレンHOMOが同位相でつながったものです。直鎖状では末端があるためMO係数が中央に集中していますが、環状では末端が無いため系全体にMO係数が均等に分布しています。末端を無くすことで単位ユニットだけでも系全体の軌道の描像を予想できます。
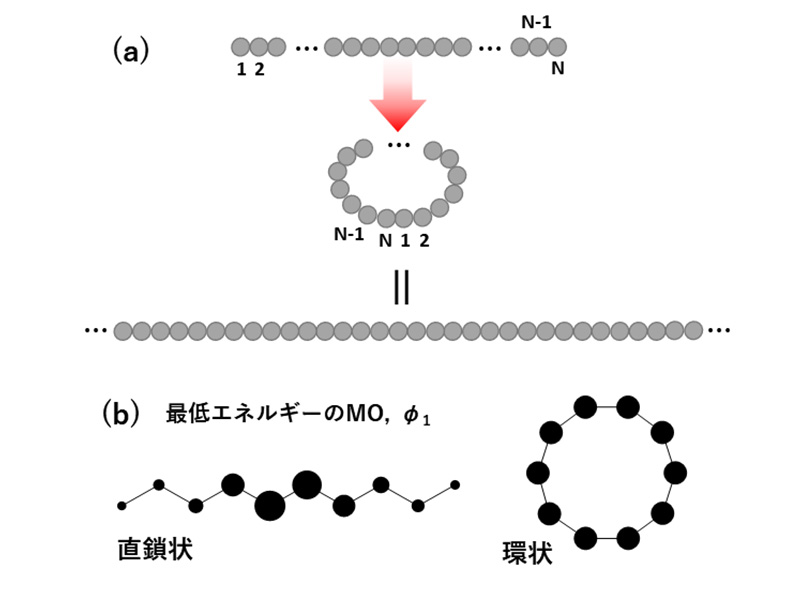 図9 (a)周期境界条件, (b)直鎖・環状ポリエンのMO
図9 (a)周期境界条件, (b)直鎖・環状ポリエンのMO
高分子をモデル化せず、大きいサイズのまま扱いたいときの最初の選択肢は量子化学計算の近似の程度を大きくすることです(
図2)。半経験的MO法であれば、かなり大きな分子まで計算可能です。π共役系に限ればSHMO法も候補です。注目部分だけを量子力学(QM)で扱い、周囲部分を古典的な分子力学(MM)で扱うQM/MM法もよく使われますが、どの範囲をQMで扱うかはモデル切り出しと同様の検討が必要です。非経験的MO法レベルで計算したいという場合には、オーダーN法という選択肢もあります。
図3のように系のサイズNに一次比例した計算コスト
O(N
1)を目指した手法で、色々なアプローチが提案されています。ただし、効率のために精度が犠牲になっていないかなど、高効率性の仕組みを良く理解した上で使用した方が良いでしょう。
おわりに
量子化学計算を高分子に適用するには、計算コスト、計算レベル、モデル化の程度など考えるべきことは多いですが、その中で、工夫や試行錯誤によって如何に知りたい情報を抽出するかが、研究者の腕の見せ所でもあります。また、量子化学計算ソフトウェアを用いてデータを得ること自体は、以前に比べれば容易になりましたが、本解説で例示したように、MO解析などを行なえば物性・現象の起源を追求することも可能です。電子レベルでの原理原則を理解できれば、これまで思いもよらなかった革新的な材料設計や創薬に繋がるかもしれません。
参考文献
1) 新版 すぐできる量子化学計算ビギナーズマニュアル」, 平尾公彦(監修), 武次徹也(編・著), 講談社 (2015)
2) 三訂 量子化学入門 (上)(下), 米澤貞次郎/永田親義/加藤博史/今村詮/諸熊奎治(著), 化学同人(1983)
3) 分子設計のための量子化学, 西本吉助/今村詮(編・著), 山口兆/山辺信一/北浦和夫(著), 講談社サイエンティフィク(1989)
4) 新しい量子化学 電子構造の理論入門 (上)(下), A.ザボ/N.S.オストランド(著), 大野公男/阪井健男/望月祐志(訳), 東京大学出版会(1987)