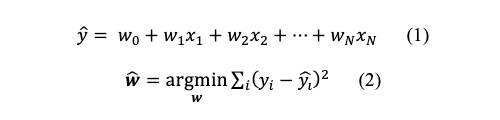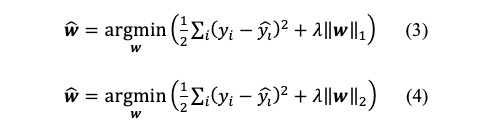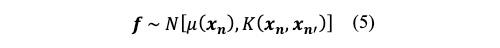執筆:林 慶浩(統計数理研究所)
高分子の特性や、所望の特性を有する高分子の構造をデータから推定する手法
高分子のデータ科学は、データから統計的な手法を用いて、高分子の構造・組成と機能・特性との関係を推定します。機械学習などの統計モデルを用いて、データから入力
xと出力
y間の関数関係
y =
f(
x) を求めることで、例えば高分子の構造
xから特性
yの予測を行うことができます。また、この関数の逆写像
x =
f -1(
y)を求めることで、所望の特性
yを有する高分子構造
xの予測を行うことにより分子設計に用いることもできます。
測定できること
物性値予測 / クラス分類予測 / スペクトル予測 / 微細構造(画像)予測 / 分子設計 / プロセス条件最適化 / 逆合成解析
原理
1. 高分子のデータ科学について
マテリアルズインフォマティクス(MI)と呼ばれる、機械学習などのデータ科学的手法を用いた材料設計における最も基本的なワークフローは、順問題と逆問題から構成されます(図1)。順問題では、入力
Sから特性
Yを予測する機械学習モデル
Y =
f(
S)を構築します。入力
Sには構造(分子構造、組成、結晶構造等)や温度、プロセス条件等が相当し、特性
Yには連続値(物性値など)、クラスラベル(層構造など)、スペクトル、画像(SEM画像など)といったものが相当します。実際には、分子の1次構造を表す文字列であるSMILESから分子記述子(あるいは単に記述子)とよばれる分子の構造的あるいは物理化学的特徴を表す数値ベクトルに変換したものを入力に用いることが最も一般的です。一方で逆問題では、順問題の機械学習モデルの逆写像
S* =
f-1(
Y*)を求めて、所望の特性
Y*を有する入力
S*の予測を行います。
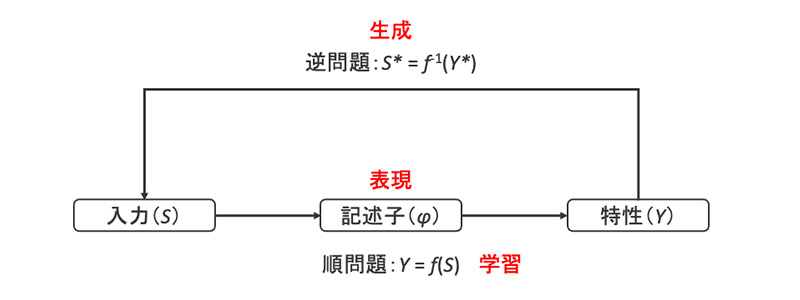 図1 マテリアルズインフォマティクスにおける順問題と逆問題
図1 マテリアルズインフォマティクスにおける順問題と逆問題
これら一連の計算は、物質や材料の“表現・学習・生成”というタスクに帰着します。一般的な機械学習のフレームワークでは、入力には要素数が固定のベクトルが用いられます。“表現”のタスクでは、高分子の化学構造や高次構造、組成などをどのような記述子という固定長ベクトルで表現するかという問題です。この記述子を入力とし、入力から特性の数学的写像
Y =
f(
S) を“学習”します。予測する特性
Yが連続値の場合は回帰タスクとなり、離散値の場合は分類タスクとなります。さらに、この機械学習モデルの逆写像を求めて、所望の特性
Y*を有する入力
S*を“生成”します。
また、データ科学を実践する上で何より重要なものはデータです。しかしながら、無機材料などの他の材料分野と比べて、高分子材料分野はデータベースが圧倒的に乏しい状況です。高分子材料の最大のデータベースは、物質・材料研究機構が開発しているPoLyInfo
1)という文献情報ベースのデータベースです。約2万骨格に対するデータが採録されていますが、ある物性値に関してのみ抽出すると数十件しかない場合も多いという状況です。また、密度汎関数法による計算値を含むデータベースとして、PolymerGenome
2)がありますが、データ数は800件程度にとどまります。加えて、いずれのデータベースも、データを一括でダウンロードできないために手作業で収集する必要があります。
2. 高分子の表現方法
化学構造式そのものはプログラム上で取り扱いにくいため、MIでは一般的に分子の化学構造はSMILESとよばれる分子の1次構造を表す文字列に変換して取り扱います。SMILESでは、原子を元素記号で表し、環、分岐、結合次数などを厳密に記述するための文法が定められています。高分子の場合、繰り返し単位をSMILES化し、繰り返し単位の両端をアスタリスクで表す例が多く見られます(図2)。ただし、この高分子の表記方法は、SMILES記法で正式に定められたものではないため、異なる表記方法を用いる場合もあります。また、ランダムコポリマーや架橋ポリマーのような1次構造を一意に定めることができない高分子構造をSMILESでは表記できないという問題があります。
.jpg) 図2 高分子構造のSMILES表現
図2 高分子構造のSMILES表現
一般的な機械学習のフレームワークでは、入力は要素数が固定の数値ベクトルであることが要請されます。そのため、SMILESの文字列を、記述子とよばれる分子の構造的あるいは物理化学的特徴を表す固定長ベクトルに変換します。最も基本的な記述子は、分子フィンガープリントです。これは、分子構造の各フラグメントの有無(バイナリ型)や頻度(カウント型)に基づき化学構造を数値化します。フィンガープリントには、事前に定義されたフラグメントを数え上げる事前定義型と、フラグメントを自動列挙する列挙型があります(図3)。事前定義型では、カルボキシル基やアミノ基といった事前に定義されたフラグメントを数え上げます。MACCS keys (166フラグメント)が代表例です。列挙型では、解析対象の分子集合からある条件を満たすフラグメントを全列挙して数え上げます。第N隣接原子までのフラグメントを全列挙するECFP (extended connectivity fingerprint)
3)が代表例です。フラグメントベースの記述子で問題になるのは、繰り返し単位の両端の処理方法です。単にH原子として扱う場合もありますが、実際の高分子鎖中に存在しないフラグメントが得られたり、繰り返し単位の連結部のフラグメントが得られないという問題があります。この問題は、繰り返し単位を環状オリゴマー化し、疑似的な無限周期構造を作ることで回避できます。
また、分子量やLogP、極性表面積といった物理化学的な数値を使用することもあります。これらの分子記述子は、RDKit(
https://www.rdkit.org/)やmordred(
https://github.com/mordred-descriptor/mordred)などのPythonライブラリを用いることで、SMILESから計算することができます。
.jpg) 図3 分子フィンガープリント
図3 分子フィンガープリント3. 機械学習モデル
入力
Sから特性
Yを予測するための機械学習アルゴリズムには様々な手法が用いられます。ここでは、いくつかの代表的な機械学習アルゴリズムに絞って解説します。
3.1.線形回帰モデル
記述子のような複数の説明変数からある特性値の回帰を行う最も単純な統計モデルは重回帰モデルです。重回帰モデルでは、特性値の推定値
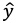
を説明変数
xの線形和とし(式1)、各データ点における特性値の観測値
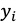
と推定値
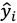
の二乗誤差を最小化する説明変数の係数
wを推定します(式2)。
この重回帰モデルでは、しばしば過学習が起きることが知られています。過学習を抑制するために、正則化項を加えた以下の式を最小化することも一般的です。
L1ノルムを正則化項に加えた式3をLasso回帰、L2ノルムを正則化項に加えた式4をRidge回帰とよびます。正則化項は係数
wが大きくなりすぎないように抑制するものです。特に、Lasso回帰では、多くの係数が0になるスパースな解が求まることが知られています。このLasso回帰の性質から、重要な記述子を抽出し知識発見に用いる例が見受けられますが、このような用途には細心の注意を払う必要があります。2つの説明変数間に相関(多重共線性)がある場合、Lasso回帰ではどちらか1つの説明変数が選択され、もう一方は係数が0になるという問題があります。これは、Lasso回帰から選択される説明変数が一意に定まらないことを意味します。そのため、Lasso回帰による変数選択を過信すると、誤った結論を導く危険性があります。
3.2.決定木
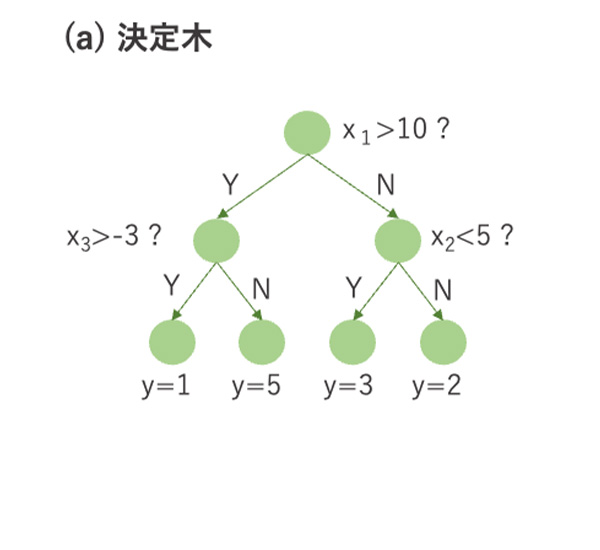
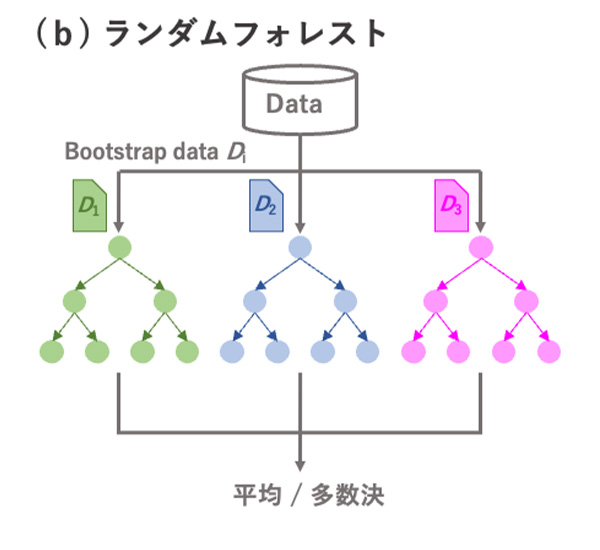
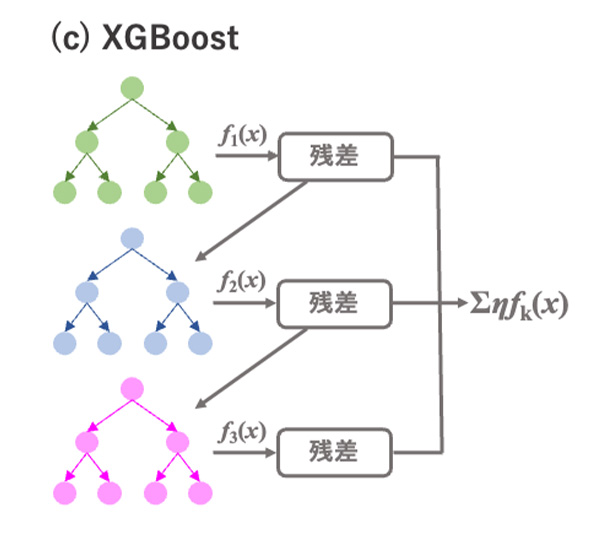
図4 決定木とランダムフォレスト, XGBoostの概略.
決定木は、木構造を用いて分類や回帰を行う機械学習の手法です。説明変数に対してあるカットオフ値に基づき複数回データを分割していきます(図4(a))。分割された各データ集合に対して分類ラベルや数値を割り当てます。条件分岐の様子を可視化できるため、結果の解釈性や説明性が高いというメリットがあります。一方で、データが少し変わると全く異なる決定木が出力されたり、予測精度が高くないという問題点があります。
決定木の予測精度を向上させるために、多数の異なる決定木を用いるアンサンブル学習を行うことが一般的です。ランダムフォレストは、ブートストラップサンプリングにより多数の異なるデータセットを用いて多数の決定木を作成します(図4(b))。また、各決定木で使用される説明変数もランダムに選択されます。これにより、異なる木構造を有する決定木が多数生成されます。回帰タスクでは多数の決定木の出力の平均値を、分類タスクでは多数決により最終的な予測値を出力します。
XGBoost(eXtreme Gradient Boosting)
4)は、ランダムフォレストとは異なる方法で決定木を用いたアンサンブル学習を行います。XGBoostでは、各決定木で目的変数ではなく残差を学習します(図4(c))。まず、最初の決定木の出力結果
f1(
x)との残差
y -
f1(
x) を計算し、次の決定木の学習で残差を小さくするための値
f2(
x)を学習し、残差
y – (
f1(
x) +
f2(
x)) を計算します。このサイクルを繰り返すことで残差を減らしていき、全ての決定木の出力値にshrinkage係数ηをかけた総和∑
kη
fk(
xi)を最終的な予測値とします。XGBoostはランダムフォレストより予測精度が向上する傾向がありますが、逐次的に学習するために計算時間がかかります。LightGBMはXGBoostとよく似たアルゴリズムでありながら、学習にかかる計算時間が短縮されており、よく使われる手法の一つとなっています。
決定木やランダムフォレストはscikit-learnで、XGBoostとLightGBMはそれぞれ同名のPythonライブラリで利用可能です。
3.3.ニューラルネットワークと転移学習
ニューラルネットワーク(NN)は、複雑な非線形関数を表現できる機械学習モデルです。NNは多数のユニットを持つ層を複数重ねた多層構造をもち、各層間のユニットを結合させたネットワーク構造となっています(図5)。各ユニットは多数の入力の線形和を活性化関数で非線形変換した値を出力します。この線形和の重み
wが学習により最適化されます。重み
wの最適化では、逆誤差伝播法により勾配を計算し、勾配法を用いて
wの更新を繰り返します。このとき、合成関数の偏微分の連鎖律より、全体の微分値は結合したユニットの微分値の積となるために、微分値が全域で1未満となる活性化関数(例えばシグモイド関数)を用いると勾配が0に近くなり(勾配消失)学習が進まなくなる問題があります。この勾配消失を抑制するために、活性化関数には、ReLU(Rectified Linear Unit)関数という、負の入力は0として、0もしくは正の入力はそのまま出力する関数がよく用いられます。
.jpg) 図5 ニューラルネットワークの概略
図5 ニューラルネットワークの概略
NNの重要な性質の一つに特徴抽出があります。NNの中間層は、入力の記述子から合成された特徴量となっています。出力に近い中間層ほど、より抽象化された特徴量となります。そして、最も出力に近い中間層では、目的変数の表現に適した特徴量がデータから自動的に抽出されることが期待されます。
NNの特徴抽出器としての性質の活用例として、転移学習とよばれる機械学習の方法論があります。転移学習は、真に解きたいタスクと関連する別のタスクのデータや学習モデルを、真に解きたいタスクに利活用する方法論全般を指します。
5)ここではNNを用いた転移学習を考えます。真に解きたいターゲットタスク(例えばある物性の実験値)はデータ量が少なく、高い予測精度をもつ機械学習モデルの構築が困難であり、一方でターゲットタスクと関連するソースタスク(例えばある物性のシミュレーション値)ではデータが多量にある状況を想定します。まず、ソースタスクのNNの学習を行います(図6)。この学習済みモデルの中間層には、ソースタスクの表現に適した特徴量が抽出されていると期待されます。そこで、この学習済みモデルのパラメータをターゲットタスクへコピーします。ターゲットタスクの最も出力側にある中間層のパラメータを初期化し、それ以外の中間層のパラメータは固定した状態で、ターゲットタスクの学習を行います。ソースタスクとターゲットタスクを表現する特徴量に共通点があれば、データ量の少ないターゲットタスクでも高い予測精度をもつモデルの構築が期待できます。
NNや転移学習は、scikit-learnやPyTorch、TensorFlow、XenonPyといったPythonライブラリで利用可能です。
.jpg) 図6 ニューラルネットワークを用いた転移学習
図6 ニューラルネットワークを用いた転移学習
無機材料などの他の材料分野と比べて、高分子材料は公開されているデータベースが圧倒的に乏しいことが、高分子のデータ科学の最大の足枷となっています。ハイスループット計算によるシミュレーションデータの大量生成と転移学習の組み合わせにより、このスモールデータ問題を克服することが試みられています。
6)
3.4.ガウス過程回帰
ガウス過程回帰モデルは、ガウス過程というランダムな関数の確率分布を利用した回帰モデルで、NNと同様に複雑な非線形関数を表現できます。
7)ガウス過程では、任意のn点の入力
xnの関数値の分布
fが、平均関数μ(
xn)、分散共分散行列
K(
xn,
xn’)のn次元ガウス分布に従います(式5)。
分散共分散行列の各要素は、カーネル関数とよばれる関数で表します。代表的なカーネル関数として、ガウスカーネルや指数カーネル、Maternカーネルがあります。ガウス過程回帰には、入力
xが似ていれば、出力
yも似ているという性質があります。カーネル関数は、この入力間の類似度を評価するものです。また、NNとの大きな違いは、出力
yの予測が確定した値でなく確率で与えられることです。その予測値はガウス分布に従い、予測の期待値と分散で与えられます。分散の値より、予測の不確かさを同時に求めることができます。
図7にガウス過程回帰の例を図示しました。ある関数系
f(
x)の真値を青い点線で示しており、そこからランダムにサンプリングしたデータ点を青でプロットしています。このデータ点を用いてガウス過程回帰を行った結果として、予測の期待値をオレンジの線で、予測の分散から計算した95%信頼区間をオレンジの幅で表しています。データ点が存在する近傍では、ガウス過程回帰の期待値は真値とよく一致しており、予測の分散も小さいことがわかります。一方で、データ点が周囲にない領域では、予測の期待値は真値から大きく外れており、その分散も大きくなっています。
ガウス過程回帰は、scikit-learnやPyTorchベースのGPyTorch、TensorFlowベースのGPflowといったPythonライブラリで利用可能です。
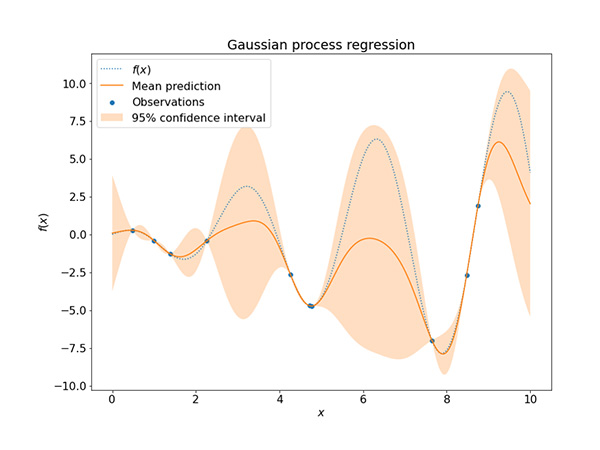 図7 ガウス過程回帰の例
図7 ガウス過程回帰の例4.逆問題と分子生成
逆問題は、構造から特性の順問題の予測モデル
Y =
f(
S) が得られたもとで、その逆写像
S =
f -1(
Y*)を求めて所望の特性
Y*を有する構造
Sを予測するタスクです。ガウス過程回帰を用いたベイズ最適化やベイズ推論に基づく逆解析がよく用いられます。
8) ここでは、MI分野でよく用いられるベイズ最適化について解説します。ベイズ最適化では、まず適当な初期データからガウス過程回帰モデルを学習します。このガウス過程回帰モデルの予測の期待値と分散を用いて定義した獲得関数を最大になるように次のサンプリング点を決めます。この獲得関数には、以下のような性質を満たすことが理想とされています。
- 良いかもしれないけど予測の不確かさが大きいところに評価を与える(探索)
- ある程度良いということがわかっているところに評価を与える(活用)
代表的な獲得関数として、Probability of Improvement (PI)やExpected Improvement (EI)、Upper Confidence Bound (UCB)があります。そして、サンプリング点のデータをなんらかの方法で取得し、ガウス過程回帰モデルを更新します。この手順を繰り返すことで、なるべく少ない試行回数で所望の特性
Y*を有する構造
Sを探索します。
図7に示したガウス過程回帰の結果を初期データ・モデルとし、ベイズ最適化を用いた最大値探索の例を図8に示しました。各サイクルにおける新たな探索点を赤い星印で示しています。サイクル1と3では予測の期待値が大きいところが選ばれ(活用)、サイクル2と4では予測の分散が大きいところが選ばれており(探索)、最終的にすべての領域で予測の期待値が真値に近くなり、分散も小さくなりました。また、最大値も3サイクル目で見つけることができています。このように、ベイズ最適化によって、効率的に所望の特性
Y*を持つ構造
Sの探索ができます。また、構造
Sのかわりにプロセス条件のパラメータを用いることで、プロセス条件最適化への応用も可能です。
.jpg) 図8 ガウス過程回帰を用いたベイズ最適化の例
図8 ガウス過程回帰を用いたベイズ最適化の例
分子設計においては、逆問題と合わせて広大な分子の設計空間を動き回ることのできる分子生成器が必要になります。これまでに、深層生成モデル
9,10)や確率的言語モデル
11)による分子生成器が開発されていますが、合成可能性の高い高分子構造の生成が難しことが大きな課題となっています。そこで、高分子の重合に用いられている重合反応の種類が比較的少ないことに着目し、重合反応ルールーベースのSMiPoly(
https://github.com/PEJpOhno/SMiPoly)
12)やOpen Macromolecular Genome(
https://github.com/TheJacksonLab/OpenMacromolecularGenome)
13)といった分子生成器が開発されています。20種類程度の重合反応のルールがあらかじめ規定されており、入力のモノマーリスト(例えば購入可能な試薬リスト)に対して重合ルールが適用できる組み合わせから高分子の分子構造を生成します。このアプローチにより、合成可能性の高い高分子構造が生成可能になっています。
参考文献
1) S. Otsuka, I. Kuwajima, J. Hosoya, Y. Xu, M. Yamazaki, In
2011 International Conference on Emerging Intelligent Data and Web Technologies 22 (2011)
2) C. Kim, A. Chandrasekaran, T. D. Huan, D. Das, R. Ramprasad,
J. Phys. Chem. C,
122, 17575 (2018)
3) D. Rogers, M. Hahn ,
J. Chem. Inf. and Model.,
50, 742 (2010)
4) T. Chen, C. Guestrin,
arXiv, (2016)
https://arxiv.org/abs/1603.02754
5) J. Jiang, Y. Shu, J. Wang, M. Long,
arXiv, (2022)
https://arxiv.org/abs/2201.05867.
6) Y. Hayashi, J. Shiomi, J. Morikawa, R. Yoshida,
npj Comput. Mater.,
8, 222 (2022)
7) 持橋 大地,大羽 成征,“ガウス過程と機械学習”,pp. 57-105,講談社 (2019)
8) 吉田 亮,劉 暢,Stephen Wu,野口 瑶,山田 寛尚,赤木 和人,大林 一平,山下 智樹,“マテリアルズインフォマティクス”,伊藤 聡 編,pp. 32-53, 共立出版 (2022)
9) R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams, A. Aspuru-Guzik,
ACS Central Science,
4, 268 (2018)
10) X. Yang, J. Zhang, K. Yoshizoe, K. Terayama, K. Tsuda,
Sci. Tech. Adv. Mater.,
18, 972 (2017)
11) H. Ikebata, K. Hongo, T. Isomura, R. Maezono, R. Yoshida,
J. Comput. Aided Mol. Des.,
31, 379 (2017)
12) M. Ohno, Y. Hayashi, Q. Zhang, Y. Kaneko, R. Yoshida,
ChemRxiv, (2023) DOI:10.26434/chemrxiv-2023-w54wn
13) S. Kim, C. M. Schroeder, N. E. Jackson,
ACS Polym. Au, (2023) DOI:10.1021/acspolymersau.3c00003